
King J, Choi G, Piret J. The etiology of Type I Diabetes and its relationship to the streptozocin family of streptomyces toxins. HPHR. 2022;63. 10.54111/0001/KKK1
The prevalence of type I diabetes continues to be a significant source of ill health, particularly in young people. The epidemiology of Type I diabetes is distinctive, with large differences among the populations of different nations. Most studies of damage to the insulin producing islet of Langerhans cells use Streptozocin-induced animal models for type I diabetes. Streptozocin (formerly called Streptozotocin) is a glucoseamine nitroso-urea produced naturally by the soil bacteria Streptomyces achromogenes. The experimental literature establishes that type I diabetes in animal models results from specific uptake of streptozocin by the GLU2 receptor on pancreatic islet cells, and subsequent toxicity. The autoimmune response appears to be a secondary response to the toxic damage to islet cells. Early reports on streptozocin as an anti-cancer agent, established that it was toxic to human pancreatic islet cells. These findings taken together raise the possibility that some fraction of human type I diabetes may be due to exposure to streptozocin or streptozocin-like compounds produced by Streptomyces species and entering the human food chain, or produced as a by- product of cooking, baking, or frying or other food processing operation.
Type I diabetes is one of the most prevalent diseases of youth in many nations, and has been increasing over the past decades,1 particularly among the young2 . The pathology of type I is characterized by loss of the ability of the pancreatic islet cells to produce adequate levels of insulin. With type I representing 10-15% of the overall incidence of diabetes, it is a major public health burden for families and communities. Among the highest incidence is in Finland, which reported 64.2cases/1000,000 in children below 15 years of age 3. In contrast the incidence is much lower in other nations.
Almost all known etiologies of disease have been explored for Type I diabetes. Below we briefly summarize the negative findings:
Type I diabetes is not transmissible, nor is there any substantial evidence for its association with distinct bacteria, fungi, or parasites. There has been significant investigation of the possible role of enteroviruses, which do influence the auto-immune response, which we discuss further below. However, the bulk of the evidence fails to establish entero- or other viruses as etiologic agents.
Extensive searches for a genetic components together with application of sophisticated genomics has identified dozens of loci at which variation influences susceptibility to diabetes 4, including loci that protect against diabetes 5. However, none of these mutations nor combinations of them can account for the prevalence of type I diabetes in particular populations or the rapid increase in recent decades. Though this may not be true for type II diabetes, for type 1 diabetes, 85-90% of people diagnosed with T1DM have no family history of the disease (TEDDY Group, Pediatric Diabetes, 2007). We agree with recent reviews concluding that the incidence of type 1 diabetes cannot be accounted for by mechanisms based on Mendelian inheritance 1. Even for those cases where there is familial occurrence – found with many diseases associated with exposure to environmental or occupational toxins, or nutritional deficiencies – this is no evidence for inheritance of gene variants transmitted from parents as the source.
Type 1 diabetes is often described as an auto-immune disease6. There is an extensive literature in animal models and in humans establishing the presence of B cells and T cells which recognize and damage islet cells7. However, it is important to distinguish autoimmune responses as secondary effects from their role as the primary etiology. It is very common, when an organ is damaged by exposure to some direct toxin or mechanical damage, for the disrupted cells and macromolecules induce an immune response, and self-tolerance is overcome. Thus in animal models of type 1 induced by feeding glycotoxins, the animals develop an auto-immune response which further damages the tissue8. However, in the control animals with the same immune system, pathology does not develop. Thus, there is little evidence that the initiation of the islet cell damage in humans or in animal models is due to an auto-immune response.
There is a considerable body of work with NOD mice, showing islet cell damage through an auto-immune pathway6. However, the existence of animal models carrying genetic defects in the immune response and showing auto-immune pathology, does not address the etiology of the disease in normal animals9. Thus feeding MPTP or Paraquat to rats induces Parkinson’s’ like disease, due to damage to the brain protein a-synuclein10,11. Rats and humans carrying mutations in the gene encoding a-synuclein also get Parkinson’s Disease, absent the toxin12. This observation does not negate the efficacy of MPTP or Paraquat in inducing PD in animals or humans with the wild type a-synuclein allele.
The pathology of type I diabetes follows form the death or the islet of Langerhans cells that normally produce insulin. This well-established pathology argues for considering Type I diabetes as due to exposure to an agent that is toxic to the islet cells. The general incidence data are probably best accounted for if there is some variation in the occupational, general, or home environment or diet that has not been identified. Two classes of such potential toxic agents have been studied – autoantibodies which attack the islet cells, and a family of glycotoxins, some of which are microbial products, and some which accumulate in many foodstuffs upon heat treatment, and are ingested13,14 . In their 2003 review, Myers, MacKay and Zimmet summarized evidence that toxic compounds produced by Streptomyces achromogenes growing n potatoes, were etiological agents of type I diabetes in humans. One particular glycotoxin, Streptozocin is widely used as an inducer of type I diabetes for animal models of human type 1, and is also used as anti-tumor agent to kill islet tumor cells.
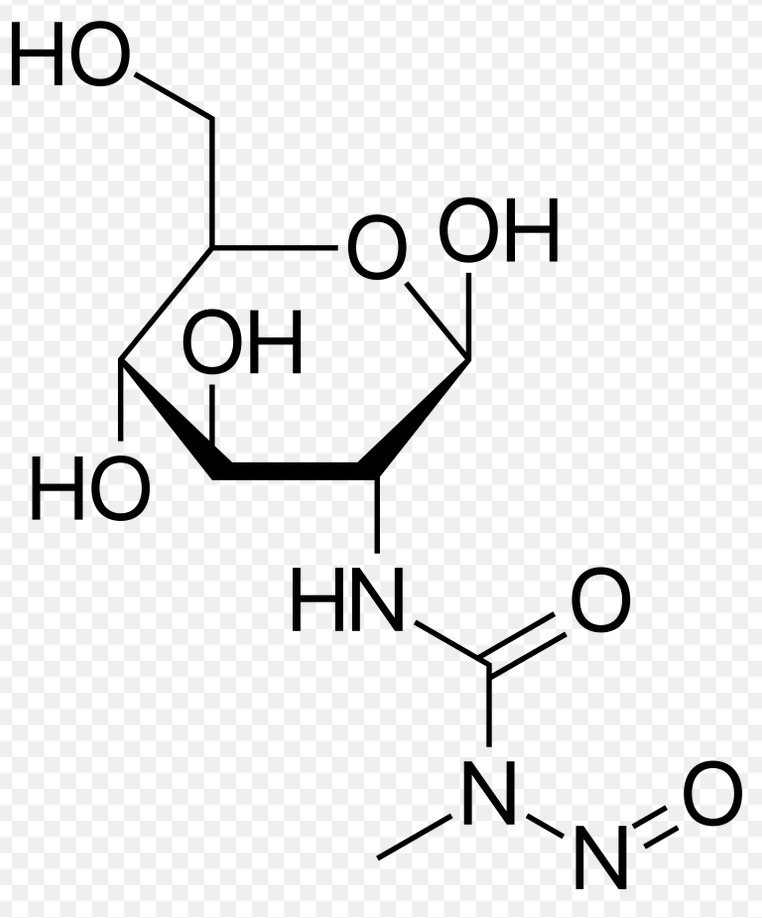
Figure 1. Chemical structure of Streptozocin
In fact there is a considerable body of evidence pointing to the presence of glycotoxins in the modern diet as influencing the incidence of type 1 diabetes 15,16. These glycotoxins modify proteins and other macromolecules generating advanced glycation end products (AGE). Nutritional origins have been investigated in different nations, but have not revealed a probable etiological agent or agents. Feeding experimental animals low AGE diets, reduces the incidence and severity of diabetic pathology. However, these experiments, though convincing, have not identified which of the multiple glycotoxins present in heated foods, are the source of the toxicity to the islet cells.
One clue to the etiology of diabetes may be from the extensive literature on the induction of human-like type 1 diabetes in animal models, and wide use of these animal models to study diabetes. The longest established of these animal models, dating from 1963, have been generated by giving rodents one or a few doses of the compound Streptozotocin or Streptozocin,17-19. Streptozocin, is produced by the widespread gram-positive bacteria Streptomyces achromogenes. The compound, originally studied as an antibiotic (“Zanosar” in the US), has a sugar moiety joined to an alkylating agent (Figure 1). As a result of the glucose moiety the compound is efficiently taken up by the GLUT2 transporter of islet cells 20,21 and then seriously damages or kills the cells. It acts as both a DNA and protein alkylating agent, though its actual mechanism of toxicity to B islet cells has not been determined. There are hundreds of well-documented studies establishing the induction of human-like diabetes after administration of streptozotocin 17.
Streptozocin is toxic to pancreatic islet cells in animals, so there is little mystery as why it should generate a deficit in insulin production.
Surprisingly, this important data on the induction of type 1 diabetes in animals, including primates, has often been ignored in the public health and epidemiology literature.
The extensive animal data raises the possibility that there is a class of compounds present or introduced into the environment which causes type I diabetes in humans by damaging pancreatic islet cells. We can identify many human diseases with such etiology – mesothelioma from asbestos; liver cancer from aflatoxin; neurological damage in Minamata disease from mercury poisoning; bladder cancer from aromatic amines; black lung from coal dust; byssinosis from cotton bract fibers.
Of particular interest are those cases in which the toxin was a product of a micro-organism present in the food chain. Some liver cancers are associated with the contamination of peanuts by aflatoxin due to the growth of the Aspergillus flavus fungus when peanuts are stored without protection from moisture22,23. In the Western Pacific neurological diseases are associated with ingestion or treatment with cycad plant extracts containing cycasin – methylazoxymethanol-B-D-glucoside 24.
Given the extensive animal experiments, it was reasonable to pursue streptozocin, or its cousins, as a potential etiological agent. Its chemistry explains its islet toxicity – a sugar so that compound is taken up by islet receptors, combined with an alkylating agent that is cytotoxic. Given that streptozotocin is toxic to islet cells in mice, rats, guinea, pigs, sheep and baboons, it is certainly reasonable that it is toxic to islet cells in humans.
The early preclinical pharmacology for streptozocin as an antibiotic included toxicity trials. In the course of these trials the effect on the pancreas of rats were noted and clearly described and reported by Nathan Rakietan, Morris Rakietan and Mareshwar Nadkrni 19, This finding was reported in chemotherapy reports and not the diabetes literature. Oncologists who learned of these animal studies showing killing of islet cells decided to test streptozocin in patients who were suffering from tumors of the pancreas and of the islet cells. They concluded that streptozocin was effective against islet cell tumors in the patients they studied, reported in a series of papers by Charles G. Moertel and colleagues et al from 1980 to 1992.25
The treatment also caused gastrointestinal distress and kidney toxicity, which tempered the author’s enthusiasm. Nonetheless, in 1982 FDA approved streptozocin for pancreatic islet cell cancer (Upjohn marketed as Zanosar).
Subsequently other oncologists dealing with endocrine tumors added streptomycin to their tests. Those papers generally do not refer back to the original animal studies on diabetogenic toxicity, but start with the Moertel anti-cancer trials. Most of these studies were done in patients with advanced tumors, otherwise they wouldn’t be included in trials of experimental anti-tumor agents. Given that the goal of the trials was to damage or kill islet tumor cells, it seems unlikely that damage to non-malignant islet cells would be monitored or reported.
Similarly, in trials focused on other advanced tumors of endocrine glands, such as adrenal tumors, it is unclear that damage to normal islet cells would have been closely monitored. The above factors make it somewhat difficult to establish or reject that streptozocin treatment of, for example, adrenal tumors, is affecting normal islet cells in those patients. In fact, the American Cancer Society description of streptozocin as an anti-tumor agent does list as a side effect altering blood sugar levels.
In summary a) Diets low in glycotoxins protect animals and humans from diabetes; b) Streptozocin, a glycotoxin formed of glucose conjugated to an alkylating agent – kills islet cells causing type I type diabetes in mice, rats, dogs, baboons, and sheep; c) in humans, it is clearly established that Streptozocin damages tumors of the islet cells. This suggests that an etiology to be explored for some fraction of human type I diabetes is exposure to streptozocin or related compound.
Three routes of possible routes of exposure to Streptozocin seem worth consideration: a) From Streptomyces growth in feedstocks or in processed foods; b) Streptozocin itself during manufacturing, distribution, or disposal; and c) Generation of a glucose-toxin conjugate during food processing or within the body similar to generate of other glycotoxins15.
Streptomyces achromogenes is a common soil bacterium widespread in the environment. Thus, the strain that makes streptozocin colonizes herbage in silos, and presumably other stored plant food stocks (potatoes, sugar beets, etc). Its prevalence could vary very widely in different geographical regions. Since Streptomyces achromogenes is not itself a human or animal pathogen, its presence is not monitored or reported through public health or agricultural oversight procedures. It is likely that other strains of Streptomyces achromogenes also produce either Streptozocin itself or a closely related compound.
Many microorganisms produce small molecules that are toxic to humans. However one class that shows similarities to the streptozocin example are the masked mycotoxins produced by Fusaria and other fungi 15,26. A variety of fungi growing on major food plants such as wheat, product secondary metabolites which are toxins to humans and animals. As part of their defense against xenobiotics the plants attach a glucose moiety, which protects them, generating “masked mycotoxins”. The extractable conjugated or non-extractable bound mycotoxins formed remain present in the plant tissue. These toxins are found in wheat, barley, oats, maize, bran flakes, beer, and cereal bars to name a few. Fusarium mycotoxins (deoxynivalenol, zearalenone, fumonisins, nivalenol, fusarenon-X, T-2 toxin, HT-2 toxin, fusaric acid) are prone to metabolization or binding by plants, but transformation of other mycotoxins by plants (ochratoxin A, patulin, destruxins) has also been described.
For many of these compounds the glucose is removed during digestion in humans, regenerating the toxin. In addition, given the presence of glucose, many of these compounds could well be taken up by islet cells, and then have the toxic moiety released by enzymatic hydrolysis within the cells.
With the growth of biomedical research, the manufacture and distribution of pure streptozocin has certainly increased. Factory and shipping employees may be routinely exposed. Students, research personnel, and hospital workers may be routinely exposed, and the excess may be entering the water supply if disposed of conventionally.
We have no reason to believe that laboratory technicians would be ingesting or inhaling significant levels of streptozocin. However, in the manufacturing, packaging and distribution centers, workers could be exposed to low doses over a long period of time. Monitoring of these facilities deserves serious consideration.
The general class of compounds, glucosamine-nitrosoureas, or glucose with some other alkylating substituent, would be expected to be toxic to islet cells given their efficient glucose uptake by glucose transporters. Such condensation products with a sugar are might generated in a particular food processing, cooking, broiling, frying, or other culturally distinct processes[13,26].
Considerable evidence already points to one of these groups, Advanced Glycation End Products 15. Though chemically somewhat diverse, these glycotoxins are produced during cooking of food stuffs, and accumulate to very different levels in different foods – for example 0.5 kU/g in whole wheat bread, but 66.5 kU/g in roasted almonds, where the units represent the levels of immunoreactivity induced. Koschinsky and coworkers established that feeding these glycotoxins was a risk factor in diabetic nephropathy27 16.
The toxic species is generated from sugar metabolism within the body. A number of studies focused more on type II diabetes have proposed that the physiological toxin is not high sugar itself, but a specific class of metabolites. Thus Advanced Glycation End products which are the toxic species, derived from high sugars 14 have been proposed as the toxic byproduct in eye disease found in diabetics.
The current major focus of type II diabetes research is altering nutrition and life cycle, lowering carbohydrate intake , reducing obesity, and increasing exercise. There can be little doubt that these programs can yield salutary results.
For type I diabetes, given the efficacy of streptozocin in inducing human-like type I diabetes in animals including primates, the biomedical community needs to seriously consider the possibility that some fraction of human type I diabetes is indeed induced by exposure to molecules that are cousins of streptozocin but more widely distributed in the human environment.
The research and training budgets of the U.S. National Institute of Diabetes and Digestive and Kidney Disease have been over one billion dollars since 2000. The largest portion of these funds has been directed to research on diabetes. However, insufficiently developed in the NIDDK research programs are serious efforts to identify potential agents in the environment or workplace that might contribute to diabetes by acting as toxins for the islets of Langerhans. Production and distribution of Streptozocin and analogues would be one place to start .
Of course, given that streptozotocin itself is natural fungal product, it may be that its cousins are also products of benign fungi or bacteria, which are in the food chain and ingested in small quantities. This would not be easy to sort out.
Nevertheless, given the burden of diabetes on the affected population and the health care system, identifying etiological agents responsible for only a small percent of cases would be a significant advance. As with lead, mercury, tobacco, asbestos and other toxins, the ability to identify them and therefore reduce human exposure, enabled to the development of widespread prevention programs. Such studies are thus worth pursuing.
The authors have no relevant financial disclosures or conflicts of interest.
The authors would like to acknowledge all members of the Philippine delegation (see full list below) who have participated in the systems thinking workshop as well as the organizers from the SingHealth Duke-NUS Global Health Institute (David Bruce Matchar, MD; John Pastor Ansah, PhD; Amina Mahmood Islam; Tan Teng Sheng Thomas) who have made the activity series possible.
Other members of the Philippine delegation: Armund Arguelles; Christian Joseph Baluyot, RND; Katrina Ceballos, MD, MBA; Ralph Emerson Degollacion; Catherine Duque-Lee MD; Alyzza Vienn Eclavea; Benjhoe Empedrado, MD; Bret Gutierrez, MPH; Miguel Angelo Mantaring, MD; Ildebrando Rodel Ruaya Jr, MHSS, RN, FISQua; Boss Sobremesana, MD; Dasha Marice Uy
Jonathan King is Prof of Molecular Biology Emeritus at MIT and co-author of more than 300 articles in peer reviewed journals on protein biochemistry, protein folding and human disease, eye lens cataract disease, and bacterial virus structure and assembly. He has a B.S. in Zoology from Yale University, Ph.D in Genetics from Caltech and was a Postdoctoral Fellow in Structural Biology with Sir Aaron Klug at the British Medical Research Council Laboratory in Cambridge UIK.
Gina Choi is an undergraduate studying Chemistry and Biological Engineering at MIT.
Jacqueline Piret is Professor of Microbiology Emeritus at Northeastern University and an authority on microbial products and byproducts in the pharmaceutical and biotech industries, including Streptomycetes. She has a BS in biochemistry from University of Wisconsin and Ph.D in Industrial Microbiology from MIT.
BCPHR.org was designed by ComputerAlly.com.
Visit BCPHR‘s publisher, the Boston Congress of Public Health (BCPH).
Email [email protected] for more information.

Click below to make a tax-deductible donation supporting the educational initiatives of the Boston Congress of Public Health, publisher of BCPHR.![]()
© 2025-2026 Boston Congress of Public Health (BCPHR): An Academic, Peer-Reviewed Journal
All Boston Congress of Public Health (BCPH) branding and content, including logos, program and award names, and materials, are the property of BCPH and trademarked as such. BCPHR articles are published under Open Access license CC BY. All BCPHR branding falls under BCPH.
Use of BCPH content requires explicit, written permission.